Several Alkene Compounds Are Given Below. Indicate if Each Compound Can Exist as Stereoisomers
Stereoisomerism
| Stereoisomers are stable compounds which have the same structural constitution, differing only in the configurational orientation of their atoms and groups in space. |
|---|
Configurational Stereoisomers of Alkenes
Equally defined in an before section, isomers are different compounds that accept the same molecular formula. When the group of atoms that make up the molecules of unlike isomers are bonded together in fundamentally dissimilar means, we refer to such compounds as constitutional isomers. For instance, the C4Height alkenes 1-butene, CH2=CHCHtwoCH3, and 2-methylpropene, (CH3)2C=CH2, are constitutional isomers. Notwithstanding, we observe that the remaining isomeric alkene, ii-butene, exists as two isomers, designated cis and trans. Concrete properties for all four isomers are given in the following table.
| Isomer | boiling pt. | melting pt. |
|---|---|---|
| 1-butene | –6.three ºC | –185 ºC |
| 2-methylpropene | –6.9 ºC | –140 ºC |
| cis-2-butene | three.7 ºC | –139 ºC |
| trans-ii-butene | 0.ix ºC | –105 ºC |
In club to sympathise how two stable isomers of 2-butene can exist, it is necessary to consider how the double bond substituents are oriented in space. The carbon-carbon double bond is formed between two sp2 hybridized carbons, and consists of 2 occupied molecular orbitals, a sigma orbital and a pi orbital. Rotation of the end groups of a double bond relative to each other destroys the p-orbital overlap that creates the pi orbital or bond. Because the pi bond has a bail free energy of roughly 60 kcal/mole, this resistance to rotation stabilizes the planar configuration of this functional group. As a event, certain disubstituted alkenes may exist as a pair of configurational stereoisomers, oft designated cis and trans.
The essential requirement for this stereoisomerism is that each carbon of the double bond must have 2 different substituent groups (one may be hydrogen). This is illustrated by the following general formulas. In the first example, the left-hand double bail carbon has 2 identical substituents (A) then stereoisomerism about the double bond is not possible (reversing substituents on the right-hand carbon gives the same configuration). In the next two examples, each double bond carbon atom has 2 different substituent groups and stereoisomerism exists, regardless of whether the two substituents on one carbon are the same as those on the other.
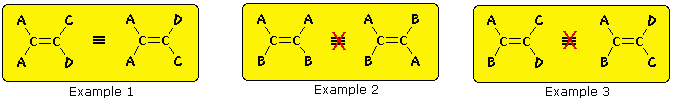
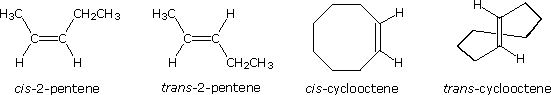
Some examples of this configurational stereoisomerism (sometimes called geometric isomerism) are shown below. Note that cycloalkenes smaller than 8 carbons cannot exist in a stable trans-configuration due to ring strain. A similar brake holds against cycloalkynes smaller than x carbons. Since alkynes are linear, there is no stereoisomerism associated with the carbon-carbon triple bond.
Nomenclature of Alkene Stereoisomers
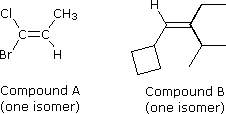 Configurational stereoisomers of the kind shown above demand an boosted nomenclature prefix added to the IUPAC name, in order to specify the spatial orientations of the groups attached to the double bond. Thus far, the prefixes cis-and trans- take served to distinguish stereoisomers; nonetheless, it is not ever clear which isomer should exist called cis and which trans. For example, consider the two compounds on the right. Both compound A (i-bromo-i-chloropropene) and compound B ( 1-cyclobutyl-2-ethyl-3-methyl-ane-butene) can be as a pair of configurational stereoisomers (one is shown). How are we to name these stereoisomers so that the configuration of each is unambiguously specified? Consignment of a cis or trans prefix to any of these isomers can simply exist done in an arbitrary fashion, so a more rigorous method is needed. A competely unambiguous system, based on a fix of group priority rules, assigns a Z (German, zusammen for together) or East (German, entgegen for reverse) to designate the stereoisomers. In the isomers illustrated in a higher place, for which cis-trans notation was adequate, Z is equivalent to cis and Eastward is equivalent to trans.
Configurational stereoisomers of the kind shown above demand an boosted nomenclature prefix added to the IUPAC name, in order to specify the spatial orientations of the groups attached to the double bond. Thus far, the prefixes cis-and trans- take served to distinguish stereoisomers; nonetheless, it is not ever clear which isomer should exist called cis and which trans. For example, consider the two compounds on the right. Both compound A (i-bromo-i-chloropropene) and compound B ( 1-cyclobutyl-2-ethyl-3-methyl-ane-butene) can be as a pair of configurational stereoisomers (one is shown). How are we to name these stereoisomers so that the configuration of each is unambiguously specified? Consignment of a cis or trans prefix to any of these isomers can simply exist done in an arbitrary fashion, so a more rigorous method is needed. A competely unambiguous system, based on a fix of group priority rules, assigns a Z (German, zusammen for together) or East (German, entgegen for reverse) to designate the stereoisomers. In the isomers illustrated in a higher place, for which cis-trans notation was adequate, Z is equivalent to cis and Eastward is equivalent to trans.
To meet how complex stereoisomeric alkenes can be named by the IUPAC organisation Click Here.
| Do Problems |
|---|
These problems exam the ability to recognize, depict and proper name alkene and cycloalkane stereoisomers
Configurational Stereoisomers of Cycloalkanes
Remember, when the group of atoms that brand upwards the molecules of unlike isomers are bonded together in fundamentally different ways, we refer to such compounds every bit constitutional isomers. For example, in the example of C5Hx hydrocarbons, well-nigh of the isomers are constitutional. Shorthand structures for five of these isomers are shown below with their IUPAC names.

Note that the fifteen atoms that make up these isomers are connected or bonded in very dissimilar ways. As is true for all constitutional isomers, each different chemical compound has a dissimilar IUPAC name. Furthermore, the molecular formula provides information about some of the structural features that must be present in the isomers (e.g. a band or a double bond). Among the formulas of the four monocyclic isomers shown hither, the last does not unambiguously designate a single unique compound. Ii isomers having the constitution of 1,2-dimethylcyclopropane exist; their backdrop, together with those of the 1,1-dimethyl isomer are listed in the post-obit tabular array.
| Isomer | boiling pt. | density |
|---|---|---|
| i,1-dimethylcyclopropane | 20 ºC | 0.662 |
| 1,two-dimethyl–isomer A | 37 ºC | 0.694 |
| 1,2-dimethyl–isomer B | 28 ºC | 0.669 |
To sympathize the origin of this isomerism it is necessary to recollect three-dimensionally, and to consider the orientation or configuration of the atoms and groups in space. The 3 carbons of the cyclopropane ring define a airplane, and the substituent atoms or groups bonded to these carbon atoms are directed in space above and beneath this plane. In the case of the 1,two-dimethyl isomers, the two methyl groups may lie on the same side of the ring, called a cis configuration, or on opposite sides, a trans configuration, every bit shown in the following diagram. From evidence that will non be described here, isomer A has the cis configuration, and isomer B the trans configuration. This configurational isomerism is unremarkably called stereoisomerization.

Drawing and Naming Cycloalkane Stereoisomers
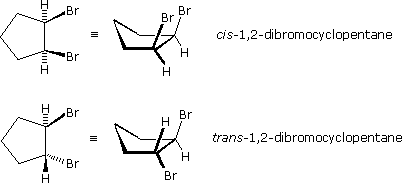 Stereoisomers are often plant in disubstituted (and college substituted) cyclic compounds. Chemists utilise heavy, wedge-shaped bonds to indicate a substituent located above the average plane of the ring (note that cycloalkanes larger than three carbons are not planar), and a hatched line for bonds to atoms or groups located below the average ring plane. As noted above, disubstituted cycloalkane stereoisomers may be designated by the nomenclature prefix cis, if the substituents are bonded on the same side of the band, or trans for substituents oriented on opposite sides of the ring. The stereoisomeric 1,2-dibromocyclopentanes shown to the right are an example.
Stereoisomers are often plant in disubstituted (and college substituted) cyclic compounds. Chemists utilise heavy, wedge-shaped bonds to indicate a substituent located above the average plane of the ring (note that cycloalkanes larger than three carbons are not planar), and a hatched line for bonds to atoms or groups located below the average ring plane. As noted above, disubstituted cycloalkane stereoisomers may be designated by the nomenclature prefix cis, if the substituents are bonded on the same side of the band, or trans for substituents oriented on opposite sides of the ring. The stereoisomeric 1,2-dibromocyclopentanes shown to the right are an example.
In general, if any two sp3 carbons in a band take 2 different substituent groups (not counting other ring atoms) stereoisomerism is possible. Four other examples of this kind of sterioisomerism in cyclic compounds are shown below.
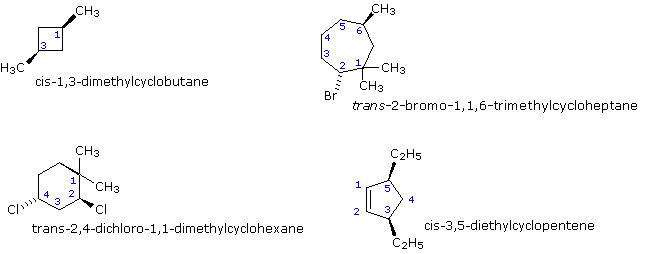
If more 2 band carbons have different substituents (not counting other ring atoms) the stereochemical notation distinguishing the diverse isomers becomes more complex.
For examples of how such compounds are named in the IUPAC system Click Hither.
Conformational Stereoisomers
Structural formulas show the manner in which the atoms of a molecule are bonded together (its constitution), but practice not mostly describe the iii-dimensional shape or configuration of a molecule. As noted above, when distinct bonding configurations permit the existence of stereoisomers, special bond notations (due east.chiliad. wedge and hatched lines) are used to distinguish the structures, and a cis or trans prefix is used to provide unique names.
In this section we shall extend our three-dimensional view of molecular structure to include compounds that unremarkably assume an assortment of equilibrating three-dimensional spatial orientations, which together narrate the same isolable chemical compound. Nosotros call these unlike spatial orientations of the atoms of a molecule that effect from rotations or twisting well-nigh unmarried bonds conformations.
In the instance of hexane, we accept an unbranched concatenation of six carbons which is often written as a linear formula: CH3CHtwoCH2CH2CH2CHiii. We know this is non strictly truthful, since the carbon atoms all have a tetrahedral configuration. The bodily shape of the extended chain is therefore zig-zag in nature. However, there is facile rotation about the carbon-carbon bonds, and the half-dozen-carbon concatenation easily coils up to assume a rather dissimilar shape. Many conformations of hexane are possible and two are illustrated below.
| Extended Chain | Coiled Chain |
|---|---|
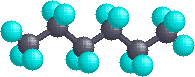 | 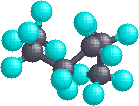 |
For an blitheness of conformational movement in hexane Click Here
1. Ethane Conformers
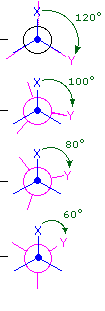
The simple alkane ethane provides a good introduction to conformational analysis. Here there is only one carbon-carbon bond, and the rotational structures (rotamers) that it may presume fall between two extremes, staggered and eclipsed. In the following description of these conformers, several structural notations are used. The first views the ethane molecule from the side, with the carbon-carbon bond being horizontal to the viewer. The hydrogens are then located in the surrounding space by wedge (in front of the plane) and hatched (behind the plane) bonds. If this structure is rotated and then that carbon #one is canted downwardly and brought closer to the viewer, the "sawhorse" projection is presented. Finally, if the viewer looks downward the carbon-carbon bond with carbon #1 in front of #ii, the Newman projection is seen.
|
| ||||||||||||||
| To see an eclipsed conformer of ethane orient itself as a Newman project, and then interconvert with the staggered conformer and intermediate conformers Click Here. | |||||||||||||||

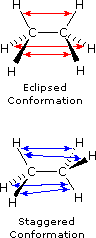
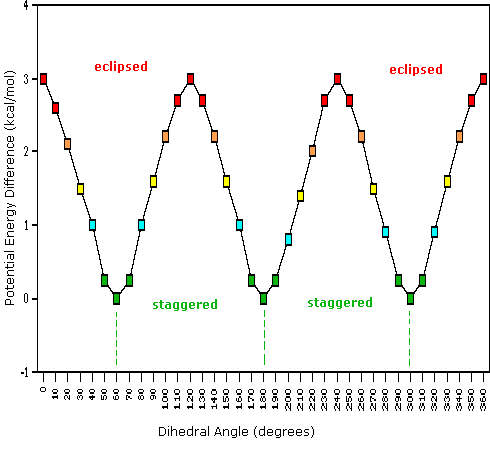
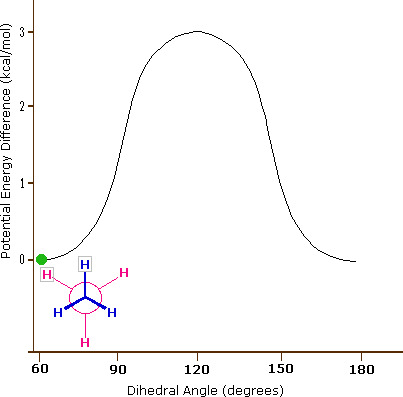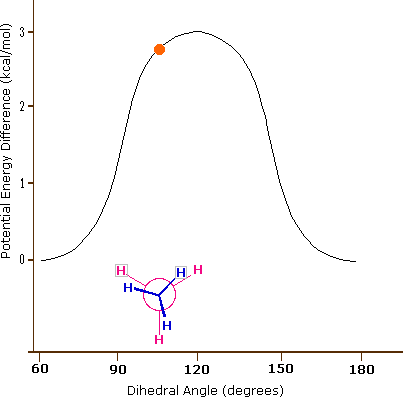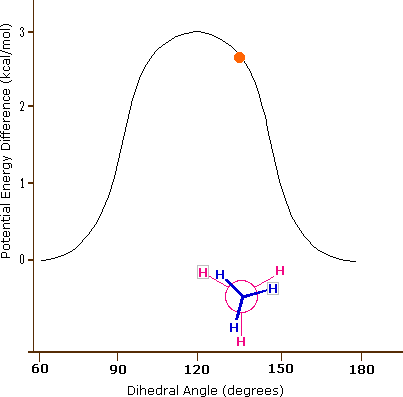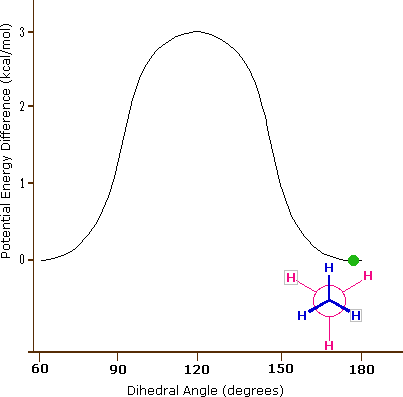
As a outcome of bond-electron repulsions, illustrated on the right above, the eclipsed conformation is less stable than the staggered conformation past roughly 3 kcal/mole. This destabilization is called eclipsing strain. The nigh severe repulsions in the eclipsed conformation are depicted past the cherry-red arrows. There are half-dozen other less stiff repulsions that are non shown. In the staggered conformation there are half-dozen equal bond repusions, four of which are shown by the blue arrows, and these are all substantially less astringent than the three strongest eclipsed repulsions. Consequently, the potential energy associated with the various conformations of ethane varies with the dihedral angle of the bonds, as shown below. Although the conformers of ethane are normally in rapid equilibrium with each other, the three kcal/mole energy difference leads to a substantial preponderance of staggered conformers (> 99.nine%) at any given time.
| Potential Energy Profile for Ethane Conformers | Dihedral Angle | |
|---|---|---|
 |
|
 |
| The above animation illustrates the human relationship betwixt ethane's potential free energy and its dihedral bending |
2. Butane Conformers
The hydrocarbon butane has a larger and more than complex set of conformations associated with its constitution than does ethane. Of detail interest and importance are the conformations produced past rotation about the central carbon-carbon bail. Amid these nosotros shall focus on two staggered conformers (A & C) and ii eclipsed conformers (B & D), shown below in several stereo-representations. As in the example of ethane, the staggered conformers are more stable than the eclipsed conformers by 2.8 to 4.5 kcal/mole. Since the staggered conformers represent the chief components of a butane sample they have been given the identifying prefix designations anti for A and gauche for C.
| Four Conformers of Butane |
|---|
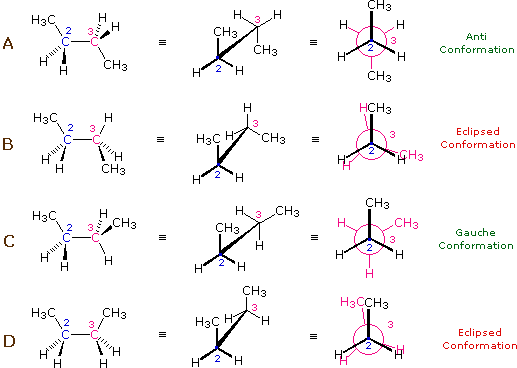 |
The post-obit diagram illustrates the modify in potential energy that occurs with rotation nigh the C2–C3 bond. The model on the right is shown in conformation D, and by clicking on any of the colored data points on the potential free energy curve, it will change to the conformer corresponding to that point. The full rotation volition exist displayed by turning the blitheness on. This model may exist manipulated past click-dragging the mouse for viewing from any perspective.
| Potential Free energy Contour for Butane Conformers | |
|---|---|
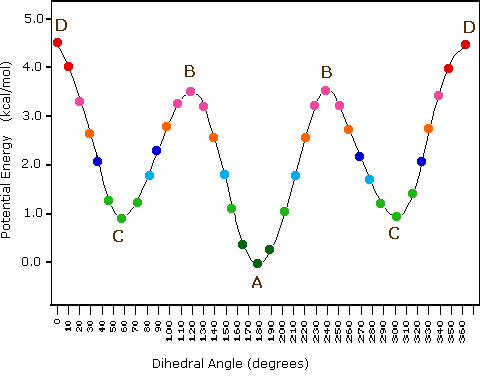 | |
| Stick Model Animation on/off |
It is useful to summarize some important aspects of conformational stereoisomerism at this time.
(ii) Specific conformers require special nomenclature terms such as staggered, eclipsed, gauche and anti when they are designated.
(3) Specific conformers may besides be designated by dihedral angles. In the butane conformers shown above, the dihedral angles formed by the 2 methyl groups near the cardinal double bond are: A 180º, B 120º, C 60º & D 0º.
(iv) Staggered conformations near carbon-carbon single bonds are more stable (have a lower potential energy) than the corresponding eclipsed conformations. The higher energy of eclipsed bonds is known as eclipsing strain.
(five) In butane the gauche-conformer is less stable than the anti-conformer by about 0.nine kcal/mol. This is due to a crowding of the ii methyl groups in the gauche structure, and is called steric strain or steric hindrance.
(vi) Butane conformers B and C have non-identical mirror image structures in which the clockwise dihedral angles are 300º & 240º respectively. These pairs are energetically the same, and have not been distinguished in the potential free energy diagram shown here.
| For a more all-encompassing discussion of rotamer analysis Click Here. |
|---|
Ring Conformations
Although the customary line drawings of simple cycloalkanes are geometrical polygons, the bodily shape of these compounds in nearly cases is very dissimilar.
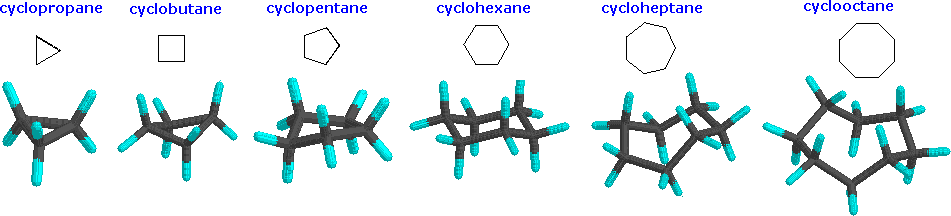
Cyclopropane is necessarily planar (flat), with the carbon atoms at the corners of an equilateral triangle. The 60º bond angles are much smaller than the optimum 109.5º angles of a normal tetrahedral carbon cantlet, and the resulting angle strain dramatically influences the chemical behavior of this cycloalkane. Cyclopropane as well suffers substantial eclipsing strain, since all the carbon-carbon bonds are fully eclipsed. Cyclobutane reduces some bond-eclipsing strain by folding (the out-of-plane dihedral angle is about 25º), only the total eclipsing and angle strain remains loftier. Cyclopentane has very little angle strain (the angles of a pentagon are 108º), only its eclipsing strain would be large (about 10 kcal/mol) if information technology remained planar. Consequently, the five-membered ring adopts non-planar puckered conformations whenever possible. Rings larger than cyclopentane would have angle strain if they were planar. All the same, this strain, together with the eclipsing strain inherent in a planar structure, tin exist relieved by puckering the ring. Cyclohexane is a good example of a carbocyclic system that virtually eliminates eclipsing and angle strain by adopting non-planar conformations, such as those shown below. Cycloheptane and cyclooctane have greater strain than cyclohexane, in large part due to transannular crowding (steric hindrance past groups on opposite sides of the ring).
ane. Some Conformations of Cyclohexane Rings
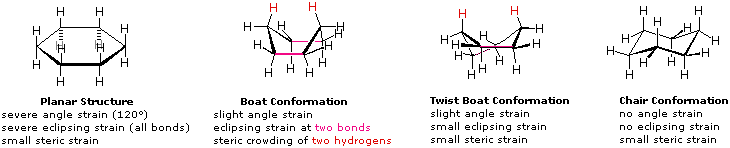
A planar construction for cyclohexane is clearly improbable. The bond angles would necessarily be 120º, x.5º larger than the ideal tetrahedral bending. As well, every carbon-carbon bond in such a structure would exist eclipsed. The resulting angle and eclipsing strains would severely destabilize this construction. If 2 carbon atoms on opposite sides of the half dozen-membered ring are lifted out of the plane of the ring, much of the angle strain tin be eliminated. This gunkhole structure withal has 2 eclipsed bonds (colored magenta in the drawing) and severe steric crowding of two hydrogen atoms on the "bow" and "stern" of the boat. This steric crowding is often called steric hindrance. By twisting the boat conformation, the steric hindrance can be partially relieved, only the twist-boat conformer notwithstanding retains some of the strains that narrate the boat conformer. Finally, past lifting one carbon atom in a higher place the ring plane and the other below the airplane, a relatively strain-gratuitous chair conformer is formed. This is the predominant structure adopted by molecules of cyclohexane.
An free energy diagram for these conformational interconversions is drawn below. The activation energy for the chair-chair conversion is due chiefly to a high energy twist-chair form (TC), in which significant angle and eclipsing strain are present. A facile twist-boat (TB)-gunkhole (B) equilibrium intervenes as one chair conformer (C) changes to the other.
Conformational Free energy Profile of Cyclohexane | ||
|---|---|---|
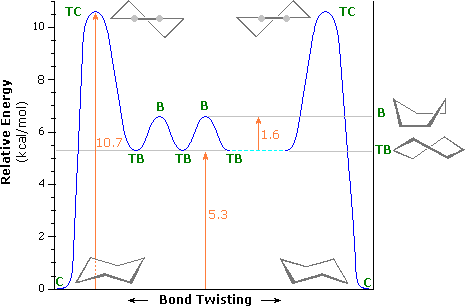 | TC = twist chair | |
| B = boat | ||
| TB = twist boat | ||
| C = chair |
These conformations may be examined every bit interactive models by Clicking Hither.
Investigations apropos the conformations of cyclohexane were initiated by H. Sachse (1890) and E. Mohr (1918), but it was not until 1950 that a total handling of the manifold consequences of interconverting chair conformers and the different orientations of pendent bonds was elucidated by D. H. R. Barton (Nobel Prize 1969 together with O. Hassel). The following discussion presents some of the essential features of this conformational assay.
On careful exam of a chair conformation of cyclohexane, we find that the twelve hydrogens are not structurally equivalent. Half-dozen of them are located well-nigh the periphery of the carbon band, and are termed equatorial. The other half dozen are oriented in a higher place and below the estimate plane of the ring (three in each location), and are termed axial because they are aligned parallel to the symmetry centrality of the ring. In the stick model shown on the left below, the equatorial hydrogens are colored blue, and the axial hydrogens are red. Since at that place are two equivalent chair conformations of cyclohexane in rapid equilibrium, all twelve hydrogens accept 50% equatorial and fifty% centric character.
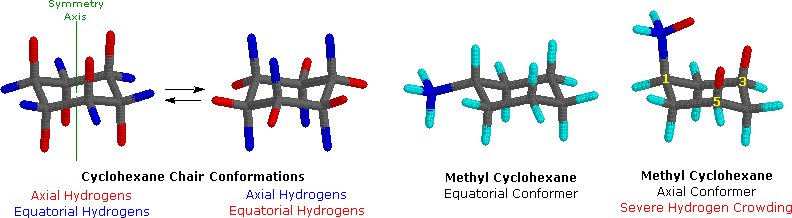
Because centric bonds are parallel to each other, substituents larger than hydrogen mostly endure greater steric crowding when they are oriented axial rather than equatorial. Consequently, substituted cyclohexanes will preferentially adopt conformations in which big substituents assume equatorial orientation. In the two methylcyclohexane conformers shown to a higher place, the methyl carbon is colored blue. When the methyl group occupies an axial position it suffers steric crowding by the two axial hydrogens located on the same side of the band. This crowding or steric hindrance is associated with the red-colored hydrogens in the structure. A careful examination of the axial conformer shows that this steric hindrance is due to 2 gauche-like orientations of the methyl group with ring carbons #iii and #v. The use of models is particularly helpful in recognizing and evaluating these relationships.
These conformations may be examined as interactive models by Clicking Here
To view an blitheness of the interconversion of cyclohexane chair conformers Clicking Here
The relative steric hindrance experienced by different substituent groups oriented in an axial versus equatorial location on cyclohexane may exist determined by the conformational equilibrium of the chemical compound. The corresponding equilibrium constant is related to the energy difference between the conformers, and collecting such information allows the states to evaluate the relative tendency of substituents to be in an equatorial or axial location. The left hand structures and tabular array in the following diagram summarize the complimentary free energy differences between equatorial and axial orientations of some simple groups. These energies are commonly reported as A values. An axial methyl grouping is hindered by two gauche butane interactions, each accounting for ca. 0.9 kcal/mol. Since an centric ethyl group may rotate so that it appears no larger than a methyl to the remaining axial hydrogens on the same side of the band, its A value is the same as methyl. Larger alkyl groups take increased A values, comensurate with increased crowding with the axial hydrogens.
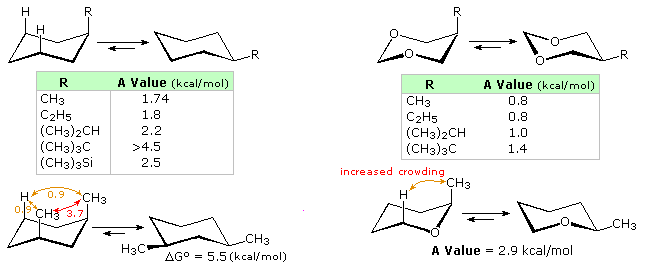
The apparent "size" of a substituent is influenced by its width and bond length to cyclohexane, as evidenced by the fact that an centric vinyl group is less hindered than ethyl, and iodine slightly less than chlorine. The trimethylsilyl group has an A value roughly half that of a tert-butyl group, reflecting the longer bond length of C–Si.
The heterocyclic compounds on the right side of the diagram illustrate the decreased centric hinderance that results from the absenteeism of nearby axial hydrogens. From the smaller merely meaning energy differences shown, it may be concluded that the steric hindrance of non-bonding electron pairs on oxygen cannot be ignored. Other factors in these cases are the shorter bond length and tighter C-O-C angle, which may act to increment hindrance, every bit shown by the lower right case.
A complete tabular array of the free energies referred to as A values may exist examined by Clicking Here.
2. Substituted Cyclohexane Compounds
Because it is then mutual amidst natural and synthetic compounds, and because its conformational features are rather well understood, we shall focus on the six-membered cyclohexane ring in this give-and-take. In a sample of cyclohexane, the ii identical chair conformers are present in equal concentration, and the hydrogens are all equivalent (50% equatorial & 50% axial) due to rapid interconversion of the conformers. When the cyclohexane ring bears a substituent, the 2 chair conformers are not the aforementioned. In one conformer the substituent is axial, in the other information technology is equatorial. Due to steric hindrance in the centric location, substituent groups prefer to be equatorial, and then that chair conformer predominates in the equilibrium.
We noted before that cycloalkanes having ii or more substituents on unlike band carbon atoms exist every bit a pair (sometimes more) of configurational stereoisomers. Now we must examine the way in which favorable ring conformations influence the backdrop of the configurational isomers. Recall, configurational stereoisomers are stable and do not easily interconvert; whereas, conformational isomers usually interconvert rapidly. In examining possible structures for substituted cyclohexanes, it is useful to follow ii principles.
(i) Chair conformations are more often than not more stable than other possibilities.
(two) Substituents on chair conformers prefer to occupy equatorial positions due to the increased steric hindrance of axial locations.
The following equations and formulas illustrate how the presence of two or more than substituents on a cyclohexane ring perturbs the interconversion of the ii chair conformers in ways that can exist predicted.
Conformational Structures of Disubstituted Cyclohexanes
| one,1-dimethylcyclohexane | 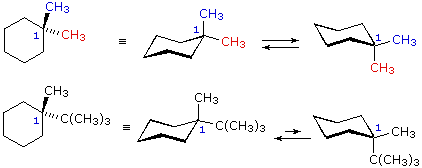 |
|---|---|
| 1-t-butyl-i-methylcyclohexane | |
| cis-1,2-dimethylcyclohexane | 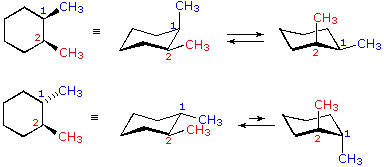 |
| trans-1,2-dimethylcyclohexane | |
| cis-1,3-dimethylcyclohexane | 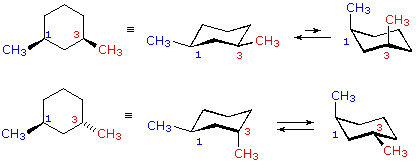 |
| trans-i,3-dimethylcyclohexane | |
| cis-1,4-dimethylcyclohexane | 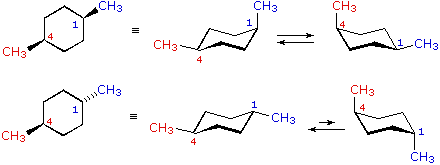 |
| trans-1,iv-dimethylcyclohexane |
In the case of 1,1-disubstituted cyclohexanes, one of the substituents must necessarily be axial and the other equatorial, regardless of which chair conformer is considered. Since the substituents are the aforementioned in i,one-dimethylcyclohexane, the two conformers are identical and present in equal concentration. In 1-t-butyl-ane-methylcyclohexane the t-butyl group is much larger than the methyl, and that chair conformer in which the larger group is equatorial will be favored in the equilibrium( > 99%). Consequently, the methyl group in this chemical compound is nearly exclusively centric in its orientation.
In the cases of 1,2-, ane,3- and 1,4-disubstituted compounds the analysis is a bit more than complex. It is always possible to have both groups equatorial, but whether this requires a cis-relationship or a trans-human relationship depends on the relative location of the substituents. Equally we count around the ring from carbon #ane to #6, the uppermost bond on each carbon changes its orientation from equatorial (or centric) to axial (or equatorial) and back. It is important to remember that the bonds on a given side of a chair band-conformation always alternate in this fashion. Therefore, it should be clear that for cis-one,ii-disubstitution, one of the substituents must be equatorial and the other centric; in the trans-isomer both may exist equatorial. Because of the alternating nature of equatorial and centric bonds, the opposite relationship is true for one,3-disubstitution (cis is all equatorial, trans is equatorial/axial). Finally, 1,4-disubstitution reverts to the 1,2-design.
The conformations of some substituted cyclohexanes may be examined every bit interactive models by Clicking Here.
Practice Problems |
|---|
These five issues concern the recognition of unlike conformations of a given ramble structure. Axial and equatorial relationships of cyclohexane substituents are also examined.
| Return to Tabular array of Contents |
|---|
![]()
Source: https://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/chapt6.htm
{kind=link}
Posting Komentar untuk "Several Alkene Compounds Are Given Below. Indicate if Each Compound Can Exist as Stereoisomers"